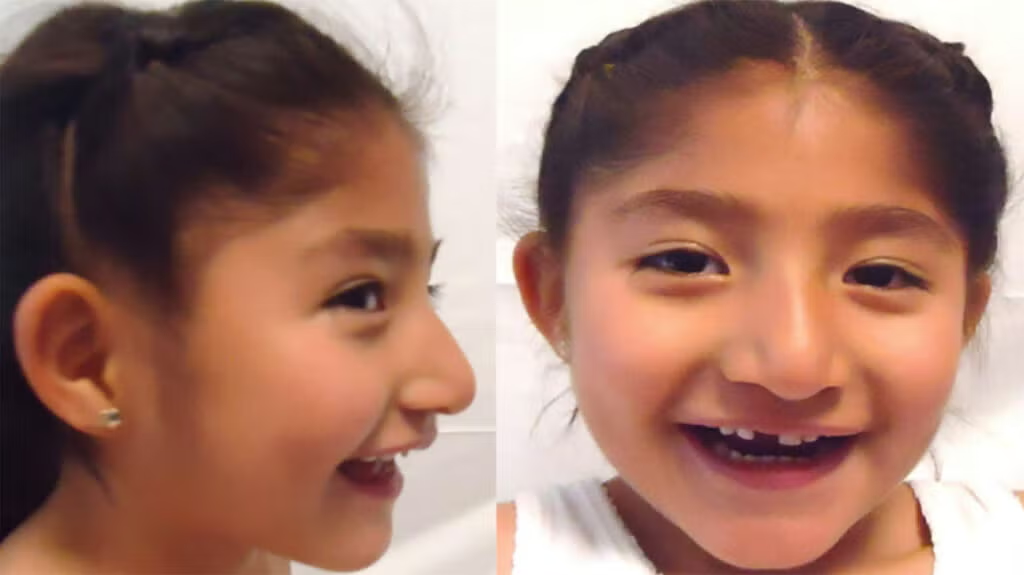
Introduction & Background of Angelman Syndrome
Angelman Syndrome (AS) is a complex, rare neurogenetic disorder that primarily affects the nervous system. It is characterized by severe developmental delays, intellectual disability, speech impairments, and a unique behavioral profile marked by frequent laughter, smiling, and an excitable personality. First described in 1965 by the British pediatrician, Dr. Harry Angelman, the condition was originally dubbed “Happy Puppet Syndrome” due to the characteristic happy demeanor and jerky movements. Today, it is recognized as a model disorder for understanding the role of genomic imprinting in human disease. While there is no cure, early diagnosis and targeted management can significantly improve the quality of life for individuals with AS and their families.
Causes of Angelman Syndrome
Angelman Syndrome is caused by the loss of function of the UBE3A gene on chromosome 15. Normally, individuals inherit one copy of this gene from each parent. Both copies are active in most of the body’s tissues, but in certain areas of the brain, only the maternal copy is active. The paternal copy is “silenced” by an imprinting process. AS occurs when the maternal copy of the UBE3A gene is missing or defective.
The primary molecular causes are:
- Deletion of Maternal Chromosome 15q11.2-q13 (~70% of cases): The most common cause, involving a small deletion on the maternal chromosome 15 that includes the UBE3A gene. This form is typically associated with more severe symptoms.
- Paternal Uniparental Disomy (UPD) (~3-5% of cases): The individual inherits two copies of chromosome 15 from the father and none from the mother.
- Imprinting Defect (~3-5% of cases): A defect in the imprinting center that incorrectly “marks” the maternal chromosome 15 as paternal, silencing the UBE3A gene.
- UBE3A Mutation (~10% of cases): A mutation within the maternal UBE3A gene itself renders it non-functional.
- Chromosome Translocation/Deletion (rare): Rare structural rearrangements affecting the critical region.
In about 10% of cases with a clinical diagnosis, the genetic cause cannot be identified with current testing.
Indications of Angelman Syndrome
Indications are the early signs that may prompt a clinical evaluation. These often become apparent between 6 and 12 months of age.
- Developmental Delay: Noticeable failure to meet milestones like sitting, crawling, or babbling on time.
- Absent or Minimal Speech: A significant lack of verbal communication, often with no words or only a few.
- Movement or Balance Disorders: Tremors, jerky limb movements, and a stiff, unsteady gait (ataxia).
- Behavioral Uniqueness: An inappropriately happy demeanor with frequent laughter, smiling, and excitability.
- Feeding Difficulties: Especially in infancy, including problems with sucking and swallowing.
Symptoms of Angelman Syndrome
The full clinical picture usually emerges in early childhood and is characterized by a consistent constellation of symptoms:
- Severe Developmental and Intellectual Disability
- Absent or Minimal Speech: Receptive language skills are stronger than expressive.
- Movement and Balance Problems: Ataxia (gait disturbance), tremors, and jerky movements.
- Behavioral Distinctiveness:
- Frequent and easily provoked laughter, smiling, and excitability.
- Hyperactive behavior.
- Short attention span.
- Seizures: Onset usually between 1-3 years of age. EEG is often abnormal, even without clinical seizures.
- Microcephaly: A small head circumference, typically by age 2.
- Sleep Disturbances: Difficulty falling and staying asleep, and decreased need for sleep.
- Fascination with Water: A strong attraction to water and crinkly items like plastic.
- Prominent Jaw and Wide-Mouthed Expression: A characteristic facial feature in many.
Prevention Strategies of Angelman Syndrome
As a genetic disorder, Angelman Syndrome cannot be prevented in the traditional sense. However, strategies focus on genetic counseling and family planning:
- Genetic Counseling: For families with a known history of AS or a previous child with AS, genetic counseling is crucial. Counselors can explain recurrence risks, which vary significantly based on the underlying genetic mechanism.
- Prenatal Diagnosis: If the genetic cause in a family is known (e.g., a UBE3A mutation or imprinting defect), prenatal testing via chorionic villus sampling (CVS) or amniocentesis can determine if the fetus is affected.
- Preimplantation Genetic Testing (PGD): For couples using in vitro fertilization (IVF), embryos can be screened for the specific genetic abnormality before implantation.
Myths and Facts About Angelman Syndrome
| Myth | Fact |
|---|---|
| Individuals with AS are always “happy.” | While they have a happy predisposition and frequent laughter, they experience the full range of human emotions, including frustration, anxiety, and anger. |
| Angelman Syndrome is a form of autism. | While there are overlapping features (e.g., speech delay, motor stereotypies), AS is a distinct genetic disorder with its own unique profile. |
| People with AS cannot learn or communicate. | They have significant intellectual disabilities but are capable of learning. They use non-verbal communication methods like gestures, communication devices, and sign language effectively. |
| It is caused by something the mother did during pregnancy. | AS is a genetic disorder, not caused by maternal actions, diet, or environmental factors during pregnancy. |
| Life expectancy is significantly shortened. | With good medical care, individuals with AS have a nearly normal life expectancy. |
Treatments and Therapy
There is no cure for Angelman Syndrome. Management is multidisciplinary and focuses on managing symptoms, maximizing development, and preventing complications.
Medication-Based Treatments
- Antiepileptic Drugs: To control seizures. Common choices include levetiracetam, clonazepam, and valproate.
- Sleep Aids: Melatonin is often used to help regulate the sleep-wake cycle.
- Medications for Behavior: Low-dose medications may be used to manage hyperactivity or anxiety if non-pharmacological interventions are insufficient.
Surgical Treatments
Surgery is not a primary treatment for AS itself but may be used for related issues, such as strabismus (eye muscle surgery) or severe gastroesophageal reflux (fundoplication).
Physical Therapy and Rehabilitation
- Physical Therapy: Improves motor skills, balance (ataxia), coordination, and strength to aid in walking and mobility.
- Occupational Therapy: Helps with fine motor skills, activities of daily living (e.g., feeding, dressing), and sensory integration.
- Speech and Language Therapy: Focuses on non-verbal communication methods (picture boards, sign language, speech-generating devices) and improving oral-motor skills for eating and possible speech sounds.
Lifestyle and Behavioral Interventions
- Structured Routines: Consistent daily schedules help reduce anxiety.
- Behavioral Strategies: Positive reinforcement is highly effective for encouraging desired behaviors.
- Sleep Hygiene: Establishing strict bedtime routines to manage sleep disturbances.
Alternative and Complementary Medicine
- Music Therapy: Can be highly effective for engagement, communication, and calming.
- Hydrotherapy: Uses water for relaxation and low-impact physical exercise.
- Massage Therapy: May help with relaxation and muscle tone.
Psychotherapy and Counseling
Primarily for parents and siblings to help them cope with the challenges of caring for a family member with a severe disability.
Immunizations and Vaccines
Individuals with AS should follow the standard immunization schedule. There is no contraindication; preventing illnesses like flu is especially important.
Stem Cell Therapy
This is currently experimental and unproven for Angelman Syndrome. There are no validated clinical trials demonstrating its safety or efficacy. It is not a standard treatment.
Gene Therapy
This is the most promising area of active research. Scientists are exploring ways to “unsilence” the paternal copy of the UBE3A gene in the brain or deliver a functional copy of the gene via viral vectors. While not yet available to patients, it represents a potential future curative pathway.
Top 20 FAQ with Answer on Angelman Syndrome
1. Is Angelman Syndrome hereditary?
It can be, but most often it is not. The vast majority of cases (especially those caused by a deletion) are sporadic (de novo). However, cases caused by UBE3A mutations or imprinting defects can have a recurrence risk of up to 50%.
2. What is the life expectancy for someone with Angelman Syndrome?
Life expectancy is nearly normal with good medical care. The condition primarily affects neurological development, not overall systemic health.
3. Can people with Angelman Syndrome talk?
Most individuals with AS do not develop functional speech. A few may learn a few words, but they rely heavily on non-verbal communication methods.
4. Can they walk?
Most children with AS learn to walk, albeit with a characteristic stiff, jerky, and unsteady gait (ataxia). The average age for walking is between 2.5 and 6 years.
5. Do individuals with AS understand what is being said to them?
Yes, their receptive language skills (understanding) are significantly stronger than their expressive skills. They often understand much more than they can express.
6. Is Angelman Syndrome similar to Autism?
There are overlaps (speech delay, motor mannerisms), but AS has a distinct genetic cause and a unique behavioral phenotype (e.g., paroxysmal laughter, ataxia) that sets it apart.
7. What causes the frequent laughter?
The exact neurological mechanism is unknown, but it is believed to be a hardwired characteristic of the disorder related to UBE3A’s role in brain function, not necessarily a reflection of an emotional state.
8. Are seizures in AS controllable?
Yes, in most cases, seizures can be well-controlled with anti-epileptic medication, though they may be difficult to manage in early childhood.
9. What is the main challenge for families?
The lifelong need for care, managing sleep disturbances, communication barriers, and ensuring the individual’s safety are among the biggest challenges.
10. Can adults with AS live independently?
No, adults with AS require supervised living and support for all aspects of daily life throughout their lives.
11. How is Angelman Syndrome diagnosed?
A clinical diagnosis is made by a geneticist or neurologist based on symptoms, which is then confirmed by genetic testing (DNA methylation test, chromosomal microarray, UBE3A sequencing).
12. Is there a prenatal test for AS?
Yes, if the genetic abnormality in the family is known, prenatal testing via CVS or amniocentesis is available.
13. What are the latest research developments?
The most exciting research is focused on gene therapy and drugs that can “unsilence” the paternal UBE3A gene.
14. Can diet or supplements help?
There is no specific “AS diet,” but a balanced diet is important. Some families report benefits from supplements, but these should always be discussed with a doctor.
15. Do they have a normal sense of pain?
Yes, but their communication difficulties may make it hard for them to express pain or discomfort, so caregivers must be vigilant.
16. How common is Angelman Syndrome?
It is considered rare, affecting approximately 1 in 12,000 to 1 in 20,000 people.
17. Can people with AS read and write?
Given the level of intellectual disability, literacy is not a typical outcome. The focus is on functional communication.
18. What happens during puberty?
Puberty occurs at the normal age. Behavioral challenges may increase during this time due to hormonal changes and frustration.
19. Are there any famous people with Angelman Syndrome?
Not in the public eye, but there are many strong and inspiring individuals and families who are advocates within the AS community.
20. Where can I find support?
Organizations like the Angelman Syndrome Foundation (USA) and Foundation for Angelman Syndrome Therapeutics (FAST) provide invaluable resources, support, and fund research.
Conclusion
Angelman Syndrome is a lifelong neurogenetic disorder that presents profound challenges for individuals and their families. Characterized by severe developmental delay, absent speech, movement disorders, and a unique behavioral phenotype, its roots lie in the disruption of the UBE3A gene on chromosome 15. While there is no cure, a multidisciplinary approach involving physical, occupational, and communication therapies can dramatically improve function and quality of life. The future holds significant promise, with groundbreaking research in gene therapy offering hope for transformative treatments. Through continued support, advocacy, and scientific discovery, the journey for those living with Angelman Syndrome can be one of progress, connection, and joy.
Find Trusted Cardiac Hospitals
Compare heart hospitals by city and services — all in one place.
Explore Hospitals
This guide explains Angelman Syndrome in a very warm and clear way, especially how developmental delays, communication challenges, and neurological features are connected, helping families understand why early intervention and supportive therapies are so important. The way you described causes, symptoms, and long-term care offers both awareness and reassurance, showing that with proper medical guidance and strong family support, individuals with Angelman Syndrome can achieve a better quality of life. A truly informative and compassionate resource for patients and caregivers.
This article explains Angelman Syndrome in a very compassionate and easy-to-understand way, especially how it connects genetics, symptoms, and daily care needs. I found the focus on early diagnosis, supportive therapies, and long-term management particularly valuable, as it gives families realistic guidance and hope. The clear, patient-focused approach makes complex medical information accessible while also highlighting the importance of awareness and continuous care for improving quality of life.