Introduction & Background of Huntington’s Disease
Huntington’s Disease (HD) is a rare, inherited, and progressive neurodegenerative disorder. It is characterized by the uncontrolled degeneration of nerve cells in specific areas of the brain, primarily the basal ganglia and cortex. This degeneration leads to a triad of severe symptoms: motor dysfunction, cognitive decline, and psychiatric disturbances. HD is caused by a single faulty gene, and it is autosomal dominant, meaning a child of an affected parent has a 50% chance of inheriting the disease. Symptoms typically appear in mid-adulthood (between ages 30-50), but can also occur earlier (Juvenile HD) or later in life. There is currently no cure for HD, but treatments are available to manage symptoms and improve quality of life.
Causes of Huntington’s Disease
The sole cause of Huntington’s Disease is a genetic mutation in the HTT gene on chromosome 4. This gene provides instructions for making a protein called huntingtin. The mutation involves an abnormal expansion of a DNA segment known as a CAG trinucleotide repeat. In a normal HTT gene, this CAG sequence repeats 10 to 35 times. In individuals with HD, the sequence is expanded, repeating 40 or more times.
- Genetic Mechanism: The expanded CAG repeat leads to the production of an abnormally long huntingtin protein. This mutant protein is toxic and gets cut into smaller, toxic fragments that clump together inside neurons, disrupting their normal functions and ultimately leading to cell death.
- Inheritance Pattern: HD is an autosomal dominant disorder. This means only one copy of the defective gene, inherited from either parent, is sufficient to cause the disease.
Indications of Huntington’s Disease
“Indications” often refer to the early signs that might prompt someone to seek medical advice. Before a formal diagnosis, early indications of HD can be subtle and easily mistaken for other conditions. These may include:
- Mild, uncontrollable fidgeting or twitching.
- Minor problems with balance or coordination (e.g., stumbling, clumsiness).
- Changes in mood, such as increased irritability, anxiety, or depression.
- Subtle shifts in personality or behavior.
- Difficulty concentrating or remembering recent events.
- A known family history of Huntington’s Disease is the strongest indication for genetic testing.
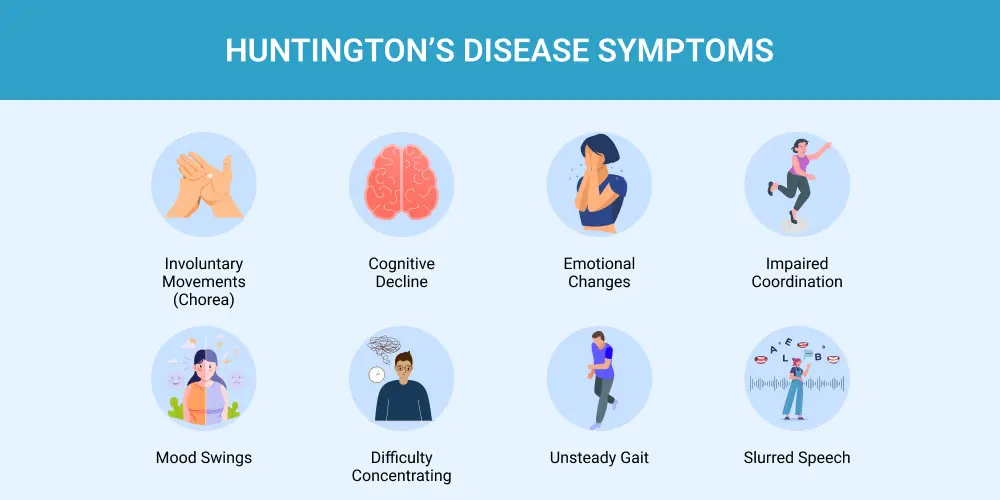
Symptoms of Huntington’s Disease
The symptoms of HD are broadly categorized into three areas, which worsen over time.
- Motor Symptoms: These involve involuntary movements (chorea) and impairments in voluntary movements.
- Chorea: Jerky, random, and uncontrollable dance-like movements.
- Dystonia: Muscle rigidity and sustained abnormal postures.
- Impaired Gait and Balance: Leading to falls.
- Difficulty with Speech and Swallowing: (Dysarthria and Dysphagia).
- Eye Movement Abnormalities: Slow or jerky eye movements.
- Cognitive Symptoms: These affect thinking and reasoning abilities.
- Slowness in processing thoughts.
- Difficulty organizing, prioritizing, or focusing on tasks.
- Lack of flexibility in thinking, or a tendency to get “stuck” on a thought/behavior (perseveration).
- Impaired judgment and reduced awareness of one’s own behaviors and abilities.
- Difficulty learning new information.
- Psychiatric Symptoms: These affect mood and behavior.
- Depression: Very common, often accompanied by insomnia, social withdrawal, and apathy.
- Obsessive-Compulsive Behaviors.
- Mania or Bipolar Disorder: In some cases.
- Irritability and Anger Outbursts.
- Anxiety.
- Psychosis: (e.g., hallucinations or delusions) is less common.
Prevention Strategies of Huntington’s Disease
As a genetic disorder, HD cannot be prevented if an individual has inherited the expanded gene. However, strategies focus on preventing its transmission to the next generation and managing its progression.
- Genetic Counseling and Testing: Individuals with a family history can undergo predictive genetic testing to learn their status. This allows for informed family planning decisions.
- Preimplantation Genetic Diagnosis (PGD): For couples where one partner has the HD gene, In Vitro Fertilization (IVF) can be used. Embryos are created and tested for the HD mutation, and only those without the expansion are implanted in the uterus.
- Prenatal Testing: During a pregnancy, procedures like chorionic villus sampling (CVS) or amniocentesis can determine if the fetus has inherited the HD gene.
- Adoption or Use of Donor Sperm/Eggs: These are alternative options to avoid passing on the gene.
Myths and Facts About Huntington’s Disease
- Myth: HD only causes involuntary movements.
- Fact: HD is a multi-faceted disease that equally devastates cognitive and psychiatric functions, which can be more debilitating than the motor symptoms.
- Myth: If your parent had HD, you will definitely get it.
- Fact: You have a 50% chance of inheriting the gene. If you do not inherit it, you cannot develop HD or pass it on.
- Myth: HD only affects older adults.
- Fact: While most common in mid-adulthood, Juvenile Huntington’s (onset before age 20) and Late-Onset HD (after 60) can occur.
- Myth: There is nothing that can be done for people with HD.
- Fact: While there is no cure, a multidisciplinary approach involving medication, therapy, and support can significantly manage symptoms and improve quality of life for many years.
- Myth: HD “skips” a generation.
- Fact: HD is a dominant disorder and does not skip generations. An unaffected individual will not pass the gene to their children. However, a person may die from another cause before their symptoms become apparent, creating the illusion of a skipped generation.
Treatments and Therapy
Management of HD requires a comprehensive, multidisciplinary approach tailored to the individual’s symptoms.
Medication-Based Treatments
- For Chorea: Tetrabenazine and Deutetrabenazine are specifically approved to reduce choreiform movements. Antipsychotics like Haloperidol or Olanzapine may also be used.
- For Psychiatric Symptoms: Antidepressants (SSRIs), mood stabilizers, and antipsychotics are used to manage depression, mood swings, and irritability.
- For Cognitive Symptoms: While no drugs are approved specifically for HD cognitive decline, some medications used for Alzheimer’s (like Donepezil) may be tried off-label, with limited evidence.
Surgical Treatments
- There are no surgical treatments to cure or slow HD. Deep Brain Stimulation (DBS) has been investigated experimentally for severe chorea and psychiatric symptoms, but it is not a standard treatment and carries significant risks.
Physical Therapy and Rehabilitation
- Aims to improve strength, flexibility, balance, and coordination to reduce fall risk and maintain mobility for as long as possible.
Lifestyle and Behavioral Interventions
- Nutrition: A high-calorie diet is often recommended, as people with HD can burn calories excessively and have difficulty eating safely. Soft or pureed foods may be needed as swallowing becomes difficult.
- Structured Routine: Maintaining a predictable daily routine can help reduce anxiety and confusion.
- Communication Strategies: Using simple sentences and non-verbal cues can aid communication as speech declines.
Alternative and Complementary Medicine
- Includes practices like mindfulness, meditation, and yoga to help manage stress and anxiety. Evidence for efficacy in HD is anecdotal.
Psychotherapy and Counseling
- Essential for patients and families to cope with the emotional and psychological impact of the diagnosis. Cognitive-behavioral therapy (CBT) can help manage depression and anxiety.
Immunizations and Vaccines
- There are no vaccines to prevent or treat HD. However, staying up-to-date on standard immunizations (like flu and pneumonia vaccines) is important to prevent infections that can be severe in someone with HD.
Stem Cell Therapy
- This is an experimental area of research. The goal is to replace damaged neurons, but it is not yet a viable or proven treatment for HD. Significant scientific and safety hurdles remain.
Gene Therapy
- This is the most promising area of research for a potential one-time treatment. Approaches like Antisense Oligonucleotides (ASOs) are being tested in clinical trials. These are designed to “silence” the mutant HTT gene, reducing the production of the toxic huntingtin protein. While not yet approved, this represents a potential future disease-modifying therapy.
Top 20 FAQ with Answers on Huntington’s Disease
- What is Huntington’s Disease?
- It is a progressive, inherited brain disorder that causes uncontrolled movements, emotional problems, and loss of thinking ability.
- What causes HD?
- A single defective gene on chromosome 4 that produces a toxic protein leading to the death of brain cells.
- Is Huntington’s Disease hereditary?
- Yes. It is passed from parent to child through an autosomal dominant pattern.
- If my parent has HD, what are my chances of getting it?
- You have a 50% (1 in 2) chance of inheriting the mutated gene.
- What are the first signs of Huntington’s Disease?
- Early signs are often subtle: mild clumsiness, mood swings, irritability, depression, and slight involuntary movements.
- At what age does HD usually appear?
- Symptoms typically begin between ages 30 and 50, but can start earlier (Juvenile HD) or later.
- Is there a cure for Huntington’s Disease?
- No, there is currently no cure. Treatments focus on managing symptoms and improving quality of life.
- How is HD diagnosed?
- A genetic blood test confirming the presence of the expanded CAG repeat in the HTT gene is the definitive diagnosis. Neurological and psychiatric evaluations are also used.
- What is the life expectancy after diagnosis?
- The average is 10 to 30 years from the onset of symptoms, often depending on the progression of symptoms and complications like pneumonia or heart failure.
- Can you test for HD before symptoms start?
- Yes, through predictive genetic testing. This is a major decision and should always be done with genetic counseling.
- What is Juvenile Huntington’s Disease?
- A form of HD where symptoms begin before age 20. It often presents differently, with rigidity, slowness, and seizures, and may progress more rapidly.
- What is chorea?
- The medical term for the involuntary, jerky, dance-like movements that are a hallmark motor symptom of HD.
- Does HD affect memory?
- Yes, it affects cognitive functions, including memory, especially the ability to learn new information and recall recent events.
- Why is depression so common in HD?
- Depression can be a direct result of the brain changes caused by HD, not just a reaction to the diagnosis. It is a core symptom of the disease.
- What is the difference between HD and Parkinson’s disease?
- Both are neurodegenerative, but they affect different brain pathways. Parkinson’s primarily causes tremors, slowness, and stiffness, while HD is characterized by chorea and has a stronger cognitive/psychiatric component. They also have different genetic causes.
- Can women with HD have children?
- Yes, but there is a 50% risk of passing the gene to each child. Genetic counseling is crucial for exploring options like PGD.
- What is the role of a genetic counselor?
- To explain the genetics, risks, and testing process; provide emotional support; and help individuals and families make informed decisions.
- How can I help a family member with HD?
- Educate yourself, be patient, offer emotional support, help with daily tasks, ensure home safety, and connect with HD support organizations.
- Are there any new treatments on the horizon?
- Yes, gene-targeting therapies like ASOs (e.g., Tominersen) are in clinical trials and hold promise for potentially slowing the disease progression.
- Where can I find support?
- Organizations like the Huntington’s Disease Society of America (HDSA) and the International Huntington Association (IHA) provide resources, support groups, and information for patients and families.
Conclusion
Huntington’s Disease is a devastating, complex condition that profoundly affects every aspect of an individual’s life and places a heavy burden on their families. Its triad of motor, cognitive, and psychiatric symptoms progresses relentlessly, and a cure remains elusive. However, significant strides have been made in understanding its genetic basis, leading to the development of promising experimental therapies aimed at the root cause. For now, a compassionate, multidisciplinary care approach is the cornerstone of management, focusing on symptom control, maintaining quality of life, and providing robust support for patients and their loved ones. Ongoing research continues to provide hope for future breakthroughs that could alter the course of this challenging disease.
Find Trusted Cardiac Hospitals
Compare heart hospitals by city and services — all in one place.
Explore Hospitals
Parkinson’s disease runs in my family, both my father and uncle passed away from it. So, when I was diagnosed, I was scared but determined not to follow the same path. For years, I tried different hospital treatments and therapies, but my condition only worsened. That changed when my neurologist recommended EarthCure Herbal Clinic (www . earthcureherbalclinic . com). I decided to try their natural treatment, and within a couple of months, my symptoms started disappearing. It’s now been three years, and I’ve had no signs of Parkinson and vertigo since. I’m truly grateful to my neurologist and to EarthCure Herbal Clinic for giving me hope and health again. I wholeheartedly recommend them (EarthCure Herbal Clinic) to anyone seeking help for Parkinson’s disease.
My husband started having symptoms of Huntington’s disease 3 weeks after his first Covid shot in June of 2021 and was at the stage where he was abusive and aggressive. I was finding it very difficult to cope. Everything was my fault. Nobody else was right except him. It was like living in another world. Doctors prescribed clonazepam to control his days and Mirapex at night to sleep. It was difficult to do anything normal; I retired in April that year and was with him 24/7. We used different supplements that didn’t work. Around 7 months ago I began to do a lot of research and came across the PD-5 protocol from Limitless Herbs Center on Google; after reading reviews, we decided to buy it. The improvement was profound; he regained the ability to walk on his own, regained his speech, sleeps soundly, and has shown no sign of hallucination. Visit their website at Limitlesshealthcenter. com
My husband started having symptoms of Huntington’s disease 3 weeks after his first Covid shot in June of 2021 and was at the stage where he was abusive and aggressive. I was finding it very difficult to cope. Everything was my fault. Nobody else was right except him. It was like living in another world. Doctors prescribed clonazepam to control his days and Mirapex at night to sleep. It was difficult to do anything normal; I retired in April that year and was with him 24/7. We used different supplements that didn’t work. Around 7 months ago I began to do a lot of research and came across the Ayurvedic PD-5 protocol from Limitless Herbs Center on Google; after reading reviews, we decided to buy it. The improvement was profound; he regained the ability to walk on his own, regained his speech, sleeps soundly, and has shown no sign of hallucination. Visit their website at Limitlesshealthcenter. com
My husband was on PD-5 formula for Huntington’s disease from Limitless Health Centre for 6 months. He sleeps soundly for 8 hours, works out frequently. The treatment relieved symptoms significantly, even better than the medications he was given. Reach them at limitlesshealthcenter. com
This guide explains Huntington’s Disease in a very clear and compassionate way, especially how movement changes, cognitive decline, and emotional symptoms are connected, helping readers understand why early diagnosis and long-term care planning are so important. The way you outlined causes, progression, and management gives families a realistic yet supportive perspective, showing that with proper medical guidance and supportive therapies, quality of life can be better managed. A truly informative and reassuring resource for anyone trying to understand this complex neurological condition.
Three years ago, my mom was battling Parkinson’s disease after suffering for over seven years. Her condition had become so severe that she was losing hope. A friend recommended Dr. Madida Sam’s herbal treatment at Earthcure Herbal Clinic, and we decided to reach out through a search of earthcureherbalclinic . c o m on internet. After starting the treatment plan, we began to see steady improvement. Today, my mom is healthy, strong, and free from her symptoms completely. We are truly grateful to Earthcure Herbal Clinic for their support and care during such a difficult time.
I was diagnosed with Parkinson’s disease four years ago. For over two years, I relied on Levodopa and several other medications, but unfortunately, the symptoms kept getting worse. The tremors became more noticeable, and my balance and mobility started to decline quickly. Last year, out of desperation and hope, I decided to try a herbal treatment program from NaturePath Herbal Clinic.
Honestly, I was skeptical at first, but within a few months of starting the treatment, I began to notice real changes. My movements became smoother, the tremors subsided, and I felt steadier on my feet. Incredibly, I also regained much of my energy and confidence. It’s been a life-changing experience I feel more like myself again, better than I’ve felt in years.If you or a loved one is struggling with Parkinson’s disease, I truly recommend looking into their natural approach. You can visit their website at www. naturepathherbalclinic .com
info@ naturepathherbalclinic .com