Introduction & Background of Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic Lateral Sclerosis (ALS), often known as Lou Gehrig’s disease, is a progressive and ultimately fatal neurodegenerative disorder. It affects the nerve cells (motor neurons) in the brain and spinal cord that control voluntary muscle movement. The term “Amyotrophic” comes from Greek: “A” means no, “Myo” means muscle, and “Trophic” means nourishment—”No muscle nourishment.” As these motor neurons degenerate and die, they stop sending messages to the muscles. The brain loses its ability to initiate and control voluntary movements, leading to muscle weakening, twitching, and wasting (atrophy). ALS is progressive, meaning the symptoms get worse over time. There is currently no cure for ALS, but treatments are available to help manage symptoms and improve quality of life.
Causes of Amyotrophic Lateral Sclerosis (ALS)
The exact cause of ALS is not fully understood. In about 90-95% of cases, there is no clear family history (sporadic ALS). In 5-10% of cases, the disease is inherited (familial ALS). Research suggests that ALS is caused by a combination of genetic and environmental factors.
- Genetics: Mutations in more than a dozen genes have been linked to familial ALS. The most common is a mutation in the C9orf72 gene. Another significant mutation occurs in the SOD1 gene.
- Glutamate Excitotoxicity: A buildup of glutamate, a chemical messenger in the brain, around nerve cells can become toxic and damage them.
- Protein Mishandling: Abnormal clumps of proteins within motor neurons can interfere with their normal function and lead to cell death.
- Oxidative Stress: An imbalance between the production of free radicals and the body’s ability to counteract their harmful effects can damage cells.
- Environmental Factors: Potential links are being studied, including exposure to toxins, viral infections, and physical trauma, but no single factor has been conclusively proven.
Indications of Amyotrophic Lateral Sclerosis (ALS)
“Indications” often refer to the early signs that prompt a medical consultation. These are the initial red flags:
- Muscle weakness in a limb, often starting in a hand, arm, or leg.
- Slurred speech or trouble swallowing.
- Muscle twitching (fasciculations) and cramps.
- Difficulty with fine motor tasks (e.g., buttoning a shirt, writing).
- Tripping or stumbling due to foot drop (weakness in ankle and leg muscles).
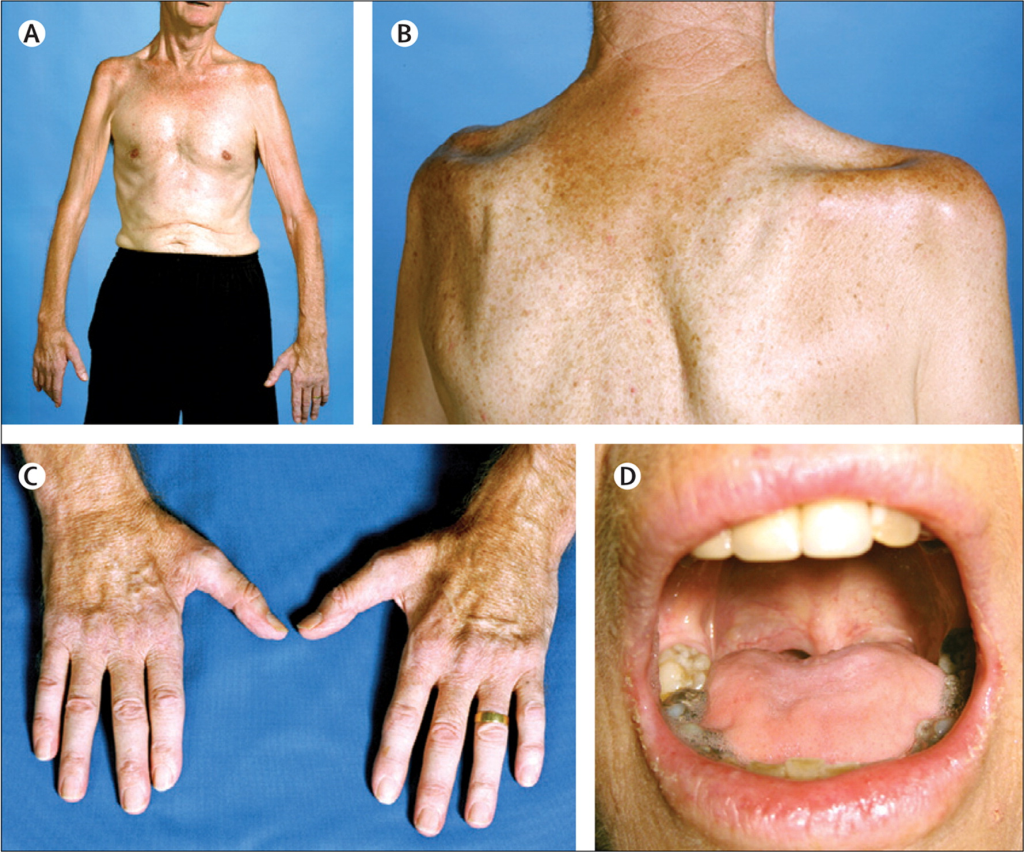
Symptoms of Amyotrophic Lateral Sclerosis (ALS)
Symptoms vary from person to person and progress over time.
Early Symptoms:
- Muscle weakness in one part of the body.
- Slurred speech (dysarthria).
- Difficulty chewing or swallowing (dysphagia).
- Muscle stiffness (spasticity), cramps, and twitches.
- Fatigue.
Progressive Symptoms:
- Weakness spreads to other parts of the body.
- Loss of ability to walk, use hands, and perform daily activities.
- Increased difficulty speaking, leading to an inability to talk.
- Breathing difficulties as muscles of the respiratory system weaken, often requiring ventilatory support.
- Unintended weight loss due to difficulty eating.
- Inappropriate emotional responses, such as laughing or crying uncontrollably (pseudobulbar affect).
- Crucially, ALS typically does not affect a person’s senses (sight, smell, taste, hearing, touch), bladder or bowel control, or cognitive function in many cases, though some may develop frontotemporal dementia (FTD).
Prevention Strategies of Amyotrophic Lateral Sclerosis (ALS)
There is no known way to prevent ALS. For individuals with a strong family history of familial ALS, genetic counseling may be recommended to understand the risks.
Myths and Facts About Amyotrophic Lateral Sclerosis (ALS)
| Myth | Fact |
|---|---|
| ALS is contagious. | Fact: ALS is not contagious. You cannot catch it from someone who has the disease. |
| Everyone with ALS experiences the same symptoms and progression. | Fact: ALS is highly variable. The progression, symptoms, and survival rate differ significantly from person to person. |
| ALS only affects motor function; the mind remains entirely intact. | Fact: While true for many, a significant portion of people with ALS experience cognitive and behavioral changes, including FTD. |
| ALS is always a painful disease. | Fact: Muscle cramping and stiffness can cause pain, but the disease itself is not characterized by pain as a primary symptom. |
| There is nothing that can be done after an ALS diagnosis. | Fact: While there is no cure, a multidisciplinary care approach can significantly improve quality of life, manage symptoms, and prolong survival. |
Treatments and Therapy
The goal of ALS treatment is to manage symptoms, slow disease progression, and maximize independence and quality of life.
Medication-Based Treatments
- Disease-Modifying Therapies:
- Riluzole (Rilutek, Tiglutik): Reduces damage to motor neurons by decreasing glutamate levels. It has been shown to prolong survival by several months.
- Edaravone (Radicava): An antioxidant that helps reduce oxidative stress, potentially slowing the decline in daily functioning.
- Sodium Phenylbutyrate/Taurursodiol (Relyvrio): Shown to slow functional decline and improve survival.
- Symptom Management: Medications for muscle cramps, spasticity, excessive saliva, pseudobulbar affect, pain, and depression.
Surgical Treatments
- Percutaneous Endoscopic Gastrostomy (PEG) Tube: A feeding tube inserted into the stomach to ensure proper nutrition and hydration when swallowing becomes difficult.
- Tracheostomy: A surgical procedure to create an opening in the neck to place a tube into the windpipe (trachea) for mechanical ventilation when breathing muscles fail.
Physical Therapy and Rehabilitation
- Physical Therapy: Maintains muscle strength and range of motion, improves cardiovascular health, and reduces spasticity and pain.
- Occupational Therapy: Helps conserve energy and maintain independence through adaptive equipment (braces, walkers, wheelchairs) and techniques.
- Speech Therapy: Teaches strategies to improve speech clarity and, when speech is lost, helps with alternative communication devices.
Lifestyle and Behavioral Interventions
- Nutritional Support: Working with a dietitian to ensure high-calorie, easy-to-swallow foods.
- Breathing Support: Use of non-invasive ventilation (BiPAP) to assist with breathing, especially during sleep.
- Energy Conservation: Pacing activities and taking frequent rests.
Alternative and Complementary Medicine
- Acupuncture: May help with pain and saliva management.
- Massage Therapy: Can help relieve muscle cramping and improve circulation.
- Mind-Body Practices: Meditation and mindfulness can help manage stress and improve psychological well-being.
Psychotherapy and Counseling
- Essential for patients and families to cope with the emotional and psychological impact of the diagnosis, including anxiety, depression, and grief.
Immunizations and Vaccines
- Getting recommended vaccines (like flu and pneumonia) is crucial to prevent respiratory infections, which can be very serious for individuals with ALS.
Stem Cell Therapy
- Status: Still experimental and not an approved treatment for ALS. Clinical trials are ongoing to see if stem cells can protect or repair motor neurons.
Gene Therapy
- Status: An emerging and highly promising area, primarily for familial ALS caused by specific genetic mutations (e.g., SOD1, C9orf72). Therapies like Tofersen (Qalsody) target the SOD1 gene mutation to slow disease progression.
Top 20 FAQ with Answers on Amyotrophic Lateral Sclerosis (ALS)
1. What is ALS?
ALS is a progressive nervous system disease that affects nerve cells in the brain and spinal cord, causing loss of muscle control.
2. Who gets ALS?
It can affect anyone, but most commonly occurs in people between the ages of 40 and 70, and is slightly more common in men than women.
3. Is ALS hereditary?
About 90-95% of cases are sporadic (no family history). 5-10% are familial (inherited).
4. What are the first signs of ALS?
Early signs often include muscle weakness in a hand, arm, or leg; slurred speech; or difficulty swallowing.
5. How is ALS diagnosed?
There is no single test. Diagnosis involves a clinical examination by a neurologist and tests like EMG, nerve conduction studies, MRI, and blood tests to rule out other conditions.
6. What is the life expectancy for someone with ALS?
The average survival time is 3 to 5 years from diagnosis, but about 10% of patients live 10 years or longer.
7. Is there a cure for ALS?
No, there is currently no cure. However, treatments can slow progression and manage symptoms.
8. Does ALS affect the mind?
It primarily affects motor neurons, but some individuals may develop cognitive and behavioral changes, including frontotemporal dementia (FTD).
9. Can you feel pain with ALS?
ALS itself is not primarily painful, but pain can result from muscle stiffness, cramps, joint immobility, or pressure on the skin.
10. Does ALS affect bladder and bowel control?
Typically, no. The nerves controlling bladder and bowel function are usually spared.
11. What is the “Ice Bucket Challenge”?
It was a 2014 social media phenomenon that raised global awareness and over $220 million for ALS research.
12. Why is it called Lou Gehrig’s disease?
It is named after Lou Gehrig, a famous American baseball player who was diagnosed with ALS in 1939.
13. Can people with ALS still communicate?
Yes. Speech therapy and augmentative and alternative communication (AAC) devices allow individuals to communicate even after they lose their voice.
14. How does ALS affect breathing?
As the muscles that control breathing weaken, it leads to shortness of breath, especially when lying down, and eventually requires ventilatory support.
15. What is the difference between ALS and MS (Multiple Sclerosis)?
Both affect the nervous system, but MS is an autoimmune disease where the immune system attacks the protective sheath around nerves, while ALS is a neurodegenerative disease that attacks the nerves themselves.
16. Can exercise help with ALS?
Yes, physical and occupational therapy are crucial to maintain function, but strenuous, exhausting exercise is not recommended.
17. What is Pseudobulbar Affect (PBA)?
A condition sometimes associated with ALS involving sudden, uncontrollable episodes of laughing or crying that don’t match the person’s actual emotions.
18. Are there any new treatments on the horizon?
Yes, research is very active. New drugs are being approved (like Relyvrio), and advanced research in gene therapy and stem cells is ongoing.
19. How can I help someone with ALS?
Offer practical and emotional support. Educate yourself about the disease, be a good listener, and assist with daily tasks as needed.
20. Where can I find support and resources?
Organizations like The ALS Association (USA), Muscular Dystrophy Association (MDA), and the International Alliance of ALS/MND Associations provide invaluable resources, support groups, and information.
Conclusion
Amyotrophic Lateral Sclerosis is a devastating disease that presents immense challenges for patients, their families, and caregivers. While a cure remains elusive, significant progress has been made in understanding its mechanisms and developing treatments that can slow its progression and manage its symptoms. A multidisciplinary approach to care, combined with ongoing, robust research into genetics, neuroprotection, and novel therapies, provides hope for better outcomes and, ultimately, a future without ALS. The global community’s support, exemplified by initiatives like the Ice Bucket Challenge, continues to be a driving force behind this vital research.
Find Trusted Cardiac Hospitals
Compare heart hospitals by city and services — all in one place.
Explore Hospitals
I was diagnosed with bulbar ALS disease in June 2024; I felt like my life had been placed on a countdown. The weakness, slurred speech, and muscle loss progressed quickly. I found the LIMITLESS HERBS CENTER ALS/MND herbal formula during one of my lowest points, and it changed everything. Within two months, I noticed more strength in my limbs, clearer speech, and a feeling of stability I didn’t have in months. This herbal formula gave me back control and hope. I feel very fortunate to have learned about the ALS/MND formula. Checkout their website www. Limitlesshealthcenter .com I’m deeply grateful for this treatment.
I was living a normal life with my family when, at 52, I began experiencing muscle stiffness and twitching. After seeing a neurologist, I was diagnosed with ALS. It was a tough reality, and as the disease progressed, I eventually lost the ability to walk and relied on a wheelchair. A friend recommended EarthCure Herbal Clinic (www. earthcureherbalclinic .com), where I began treatment under Dr. Madida Sam. After about three months, I noticed significant improvements, less stiffness, fewer symptoms, and I was able to walk distances again.
This ALS overview feels genuinely helpful because it explains the condition in a clear, step-by-step way—what ALS is, how symptoms typically show up, and why early neurological evaluation and supportive care matter so much. I also liked the balanced focus on management (speech/swallow support, breathing care, physiotherapy, nutrition, and mental health) because it reminds readers that even when a disease is serious, the right care team can protect comfort, function, and dignity for longer. A strong, compassionate resource for patients and families trying to understand what comes next.