Introduction & Background of Fabry Disease
Fabry Disease is a rare, inherited genetic disorder that affects the body’s ability to break down a specific type of fat. It is classified as a lysosomal storage disease. Lysosomes are the “recycling centers” within our cells, containing enzymes that break down waste and complex molecules. In Fabry Disease, there is a deficiency or malfunction of an enzyme called alpha-galactosidase A (α-Gal A). This enzyme’s job is to break down a fatty substance known as globotriaosylceramide (GL-3 or Gb3). When the enzyme is missing or not working correctly, GL-3 accumulates to toxic levels inside the lysosomes of cells throughout the body, particularly in the blood vessels, kidneys, heart, and nervous system. This progressive build-up causes the wide range of symptoms associated with the disease.
Causes of Fabry Disease
Fabry Disease is caused by a mutation (or defect) in the GLA gene located on the X chromosome. This gene provides the instructions for making the α-Gal A enzyme. The inheritance pattern is X-linked. This means:
- Males (XY) inherit one X chromosome. If their single X chromosome has the defective gene, they will have little to no enzyme activity and will typically experience severe symptoms. This is called classic Fabry Disease.
- Females (XX) inherit two X chromosomes. If one X chromosome has the defective gene, they are considered “carriers.” However, due to a process called X-chromosome inactivation, the expression of the disease in females can be highly variable. They may be asymptomatic, have mild symptoms, or, in some cases, be as severely affected as males. This is a key reason why Fabry Disease was long considered a “male-only” disorder, which is a myth.
Indications of Fabry Disease
The indications for testing and diagnosing Fabry Disease often arise from a combination of symptoms, especially when they appear in childhood or early adulthood. Key indications include:
- A family history of Fabry Disease or unexplained early heart, kidney, or stroke events in the family.
- Chronic pain in the hands and feet (acroparesthesia) not explained by other conditions.
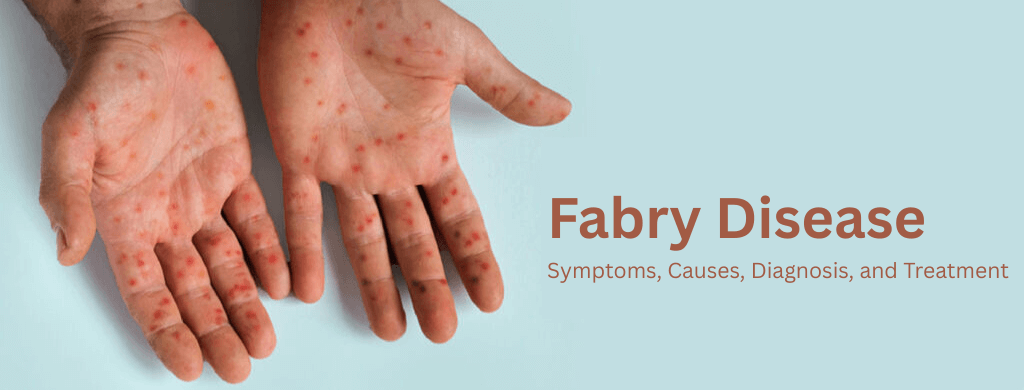
- Clusters of small, dark red spots (angiokeratomas) on the skin.
- Reduced or absent sweating (hypohidrosis or anhidrosis).
- Cloudiness of the front part of the eye (corneal opacity), visible only by slit-lamp examination.
- Unexplained kidney impairment or heart disease (especially left ventricular hypertrophy) in a young person.
Symptoms of Fabry Disease
Symptoms can begin in childhood and worsen over time, varying significantly between individuals.
Early Symptoms (often in childhood/adolescence):
- Neuropathic Pain: Burning, tingling, and excruciating pain in the hands and feet (acroparesthesia). This can be constant or occur as “Fabry crises” – episodes of severe pain triggered by fever, fatigue, or stress.
- Gastrointestinal Issues: Abdominal pain, cramping, diarrhea, nausea, and feeling full quickly after eating.
- Hypohidrosis/Anhidrosis: Inability to sweat, leading to heat intolerance and exercise fatigue.
- Angiokeratomas: Dense clusters of small, dark red, benign skin lesions, typically found between the belly button and knees.
- Corneal Verticillata: A whorl-like clouding of the cornea that does not affect vision.
Later-Stage/Organ-Specific Symptoms (often in adulthood):
- Kidney Damage: Protein in the urine (proteinuria), progressive kidney failure often requiring dialysis or transplant.
- Heart Complications: Enlarged heart (cardiomyopathy), irregular heartbeats (arrhythmias), heart valve problems, and increased risk of heart attacks.
- Neurological Complications: Increased risk of transient ischemic attacks (TIAs) and strokes, even in young people.
- Hearing Loss: Tinnitus (ringing in the ears) and sudden or progressive hearing loss.
- Psychological Impact: Depression, anxiety, and reduced quality of life due to chronic pain and the burden of a progressive disease.
Prevention Strategies of Fabry Disease
As a genetic condition, Fabry Disease itself cannot be “prevented” in the traditional sense. However, the following strategies can prevent or delay its severe complications:
- Genetic Counseling and Testing: For individuals with a family history, genetic counseling can assess the risk of passing the disease to children. Prenatal testing (like chorionic villus sampling or amniocentesis) is available.
- Family Screening: Once one person is diagnosed, it is crucial to screen other family members to identify affected individuals early, allowing for timely intervention.
- Early Diagnosis and Proactive Management: The most critical form of prevention is early diagnosis through newborn screening or family testing. Starting treatments before significant organ damage occurs can prevent or slow the progression of kidney, heart, and cerebrovascular complications.
Myths and Facts About Fabry Disease
| Myth | Fact |
|---|---|
| Fabry Disease only affects males. | While males are often more severely affected, females who are carriers can also exhibit symptoms ranging from mild to severe. |
| It’s only a pain disorder. | The pain is a major early symptom, but Fabry is a multi-systemic disease that can lead to life-threatening kidney, heart, and brain complications. |
| There is no treatment for Fabry Disease. | There are several approved treatments, including Enzyme Replacement Therapy (ERT) and Chaperone Therapy, that can manage the disease and slow its progression. |
| If you don’t have skin lesions, you don’t have Fabry. | Not all individuals with Fabry develop visible angiokeratomas. Diagnosis should not be ruled out based on their absence. |
| Life expectancy is always short. | With modern treatments and proactive management of organ complications, life expectancy for people with Fabry Disease has significantly improved. |
Treatments and Therapy
The goal of treatment is to replace the missing enzyme, manage symptoms, and slow the progression of organ damage.
Medication-Based Treatments
- Enzyme Replacement Therapy (ERT): This is the cornerstone of treatment. It involves intravenous (IV) infusions of a recombinant (lab-made) form of the α-Gal A enzyme (agalsidase alfa or agalsidase beta) every two weeks. ERT helps clear the accumulated GL-3 from cells.
- Chaperone Therapy (Oral): For individuals with specific “amenable” GLA mutations, a drug called migalastat (Galafold) can be used. It works by stabilizing the faulty enzyme, helping it fold correctly and function better within the lysosomes.
- Symptom-Specific Medications: Pain is managed with medications like anticonvulsants (e.g., carbamazepine, gabapentin) or antidepressants (e.g., amitriptyline). Medications for high blood pressure (ACE inhibitors, ARBs) are used to protect the kidneys and heart.
Surgical Treatments
Surgery is not a treatment for Fabry itself but may be necessary for its complications, such as:
- Kidney Transplant: For patients with end-stage renal disease.
- Pacemaker or Implantable Cardioverter-Defibrillator (ICD): To manage life-threatening arrhythmias caused by Fabry-related heart damage.
Physical Therapy and Rehabilitation
Can help maintain joint mobility, muscle strength, and overall function, especially if the disease has caused physical limitations.
Lifestyle and Behavioral Interventions
- Diet: A heart-healthy and kidney-protective diet (low in salt, saturated fat, and, in later stages, low in protein and potassium) is recommended.
- Hydration: Staying well-hydrated is crucial, especially for those with difficulty sweating.
- Pain Management: Avoiding triggers for pain crises (like extreme temperatures and stress) is important.
- Exercise: Regular, moderate exercise as tolerated to maintain cardiovascular health.
Alternative and Complementary Medicine
Some patients find relief with techniques like acupuncture, meditation, or yoga for pain and stress management. These should be used as complements to, not replacements for, standard medical care.
Psychotherapy and Counseling
Essential for addressing the psychological impact of living with a chronic, progressive disease. It can help manage depression, anxiety, and the emotional burden on both patients and their families.
Immunizations and Vaccines
Staying up-to-date with vaccinations, including the annual flu shot and COVID-19 vaccines, is important to prevent infections that could trigger a Fabry crisis or complicate the disease.
Stem Cell Therapy
While theoretically promising, hematopoietic stem cell transplantation is not a standard treatment for Fabry Disease due to significant risks and is generally not recommended.
Gene Therapy
This is an emerging and highly promising area of research. The goal of gene therapy is to introduce a functional copy of the GLA gene into the patient’s cells, enabling them to produce their own working enzyme. Several clinical trials are underway, offering hope for a potential one-time, curative treatment in the future.
Top 20 FAQ with Answers on Fabry Disease
1. Is Fabry Disease contagious?
No, it is an inherited genetic disorder and cannot be caught from another person.
2. How is Fabry Disease diagnosed?
Diagnosis involves measuring α-Gal A enzyme activity in blood (especially informative in males) and confirmed by genetic testing to identify the GLA gene mutation.
3. At what age do symptoms usually start?
Early symptoms like pain and GI issues often begin in childhood, typically between ages 4 and 10.
4. Can women have Fabry Disease?
Yes. Female carriers can have symptoms ranging from none to very severe, affecting the heart, kidneys, and brain.
5. What is the life expectancy for someone with Fabry?
Historically, it was reduced. With modern ERT and proactive care, many patients can live into their 60s, 70s, or longer, though this varies individually.
6. Is there a cure for Fabry Disease?
There is currently no cure, but treatments like ERT and chaperone therapy can effectively manage the disease and slow its progression.
7. Can Fabry Disease skip a generation?
No. As an X-linked disorder, the mutated gene is passed every generation, but symptoms may be so mild in a female that she is unaware she has the disease, making it appear to have skipped.
8. What is a “Fabry crisis”?
It refers to episodes of severe, debilitating pain in the hands and feet, often triggered by fever, stress, or temperature changes.
9. Can people with Fabry have children?
Yes. A affected father will pass the mutated gene to all of his daughters (who will be carriers) but none of his sons. A carrier mother has a 50% chance of passing the gene to each child, regardless of gender.
10. Why is it important to avoid getting overheated?
Because many patients cannot sweat effectively, they are at high risk for heat exhaustion and heat stroke.
11. Does Fabry Disease affect the brain?
Yes, it increases the risk of strokes and TIAs due to the accumulation of GL-3 in the small blood vessels of the brain.
12. Is pain the only symptom?
No, it is a multi-system disease that can affect the skin, eyes, gastrointestinal tract, kidneys, heart, and brain.
13. What is the difference between classic and late-onset Fabry?
Classic Fabry presents with early, multi-system symptoms in childhood/teen years. Late-onset (or atypical) Fabry presents later in life, often with isolated heart or kidney problems.
14. How effective is Enzyme Replacement Therapy (ERT)?
ERT is very effective at clearing GL-3 from cells and has been shown to stabilize kidney function, reduce heart size, and improve quality of life, especially when started early.
15. Are the treatments covered by insurance?
ERT and chaperone therapy are very expensive, but they are typically covered by most insurance plans, including Medicaid, given that Fabry is a life-threatening condition.
16. Can diet cure Fabry Disease?
No, but a healthy diet tailored to protect the kidneys and heart is a vital part of comprehensive management.
17. What specialist should a patient see?
Care is typically managed by a team, including a geneticist, nephrologist (kidney), cardiologist (heart), neurologist (brain), and pain specialist.
18. Is Fabry Disease fatal?
If left untreated, the complications (kidney failure, heart disease, stroke) can be life-threatening. With treatment, the prognosis is much improved.
19. Can you test for Fabry Disease before birth?
Yes, through prenatal diagnostic tests like chorionic villus sampling (CVS) or amniocentesis if the family mutation is known.
20. What is the most promising research for Fabry?
Gene therapy is the most promising area, with the potential to provide a long-lasting or permanent solution by correcting the underlying genetic defect.
Conclusion
Fabry Disease is a complex, multi-systemic disorder rooted in a genetic enzyme deficiency that disrupts fat metabolism. While it presents significant challenges through chronic pain and the potential for severe damage to vital organs, the landscape for patients has transformed dramatically. Advances in understanding its X-linked inheritance, the development of targeted therapies like Enzyme Replacement and Chaperone Therapy, and a growing emphasis on early diagnosis and comprehensive care have turned what was once a devastating diagnosis into a manageable chronic condition. Ongoing research, particularly in the field of gene therapy, continues to offer hope for even more effective treatments in the future, aiming to improve the quality and longevity of life for all individuals and families affected by Fabry Disease.
Find Trusted Cardiac Hospitals
Compare heart hospitals by city and services — all in one place.
Explore Hospitals
This article offers a very clear and comprehensive overview of Fabry disease, making a complex genetic metabolic disorder easier to understand for both medical professionals and general readers. I appreciate how it breaks down the underlying cause—GLA gene mutations affecting fat metabolism—and explains the wide range of symptoms from pain and skin changes to kidney and cardiac involvement. The discussion of diagnosis, enzyme replacement therapy, and other treatment options is particularly helpful for patients and caregivers seeking guidance. Articles like this play an important role in raising awareness about rare diseases and empowering readers with knowledge that can lead to earlier detection and better long‑term care outcomes.