Introduction & Background of Pompe Disease
Pompe disease is a rare, inherited genetic disorder that results in the progressive weakening of muscles throughout the body. It is classified as a Lysosomal Storage Disease (LSD). In healthy individuals, lysosomes within cells act as “recycling centers,” breaking down complex molecules. In Pompe disease, a deficiency of the enzyme acid alpha-glucosidase (GAA) prevents the breakdown of a complex sugar called glycogen. This leads to the accumulation of glycogen within lysosomes, primarily in muscle cells, which damages and weakens them. The disease is named after the Dutch pathologist, Dr. J.C. Pompe, who first described it in 1932.
Causes of Pompe Disease
Pompe disease is caused by mutations in the GAA gene, which provides the instructions for making the acid alpha-glucosidase enzyme. This genetic defect is inherited in an autosomal recessive pattern, meaning an individual must inherit two faulty copies of the gene (one from each parent) to develop the disease. Parents who each carry one mutated gene are called “carriers” and typically do not show symptoms themselves.
Indications of Pompe Disease
The primary indication of Pompe disease is the abnormal and excessive accumulation of glycogen within the lysosomes of muscle cells. This pathological hallmark is what directly leads to the clinical signs and symptoms of the disease. It can be observed through muscle biopsy or inferred through diagnostic testing.
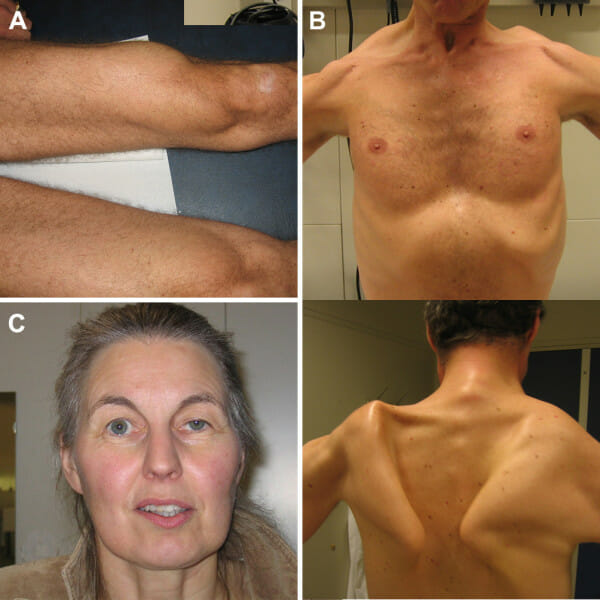
Symptoms of Pompe Disease
Symptoms vary widely in age of onset and severity, leading to two main forms, though the disease is now seen as a spectrum.
Infantile-Onset Pompe Disease (IOPD):
- Profound muscle weakness (a “floppy baby” appearance or hypotonia)
- Poor feeding and difficulty swallowing
- Failure to gain weight and grow (failure to thrive)
- Enlarged tongue (macroglossia)
- Enlarged heart (cardiomegaly) and liver (hepatomegaly)
- Respiratory difficulties and frequent lung infections
- Hearing loss
Late-Onset Pompe Disease (LOPD):
- Progressive muscle weakness, starting in the legs and trunk
- Difficulty climbing stairs, rising from a chair, or walking
- Respiratory insufficiency, especially during sleep
- Scoliosis (curvature of the spine)
- Fatigue
- Lower back pain
- Difficulty breathing
Prevention Strategies of Pompe Disease
As a genetic disorder, Pompe disease itself cannot be prevented. However, the following strategies can prevent or manage its complications:
- Genetic Counseling and Carrier Testing: For individuals with a family history of Pompe disease, genetic counseling and testing can assess the risk of passing the condition to their children.
- Newborn Screening: Many regions have added Pompe disease to their newborn screening panels. Early detection allows for prompt treatment, which can significantly improve outcomes.
- Prenatal Testing: For at-risk pregnancies, procedures like chorionic villus sampling (CVS) or amniocentesis can diagnose Pompe disease in the fetus.
Myths and Facts About Pompe Disease
- Myth: Pompe disease is contagious.
- Fact: It is a genetic disorder and cannot be spread from person to person.
- Myth: Only infants get Pompe disease.
- Fact: While a severe form appears in infancy, late-onset forms can present at any age, from childhood to late adulthood.
- Myth: There is no treatment for Pompe disease.
- Fact: Enzyme Replacement Therapy (ERT) is a specific treatment that can slow the progression of the disease and improve muscle strength and function.
- Myth: If you have muscle weakness, it must be Pompe disease.
- Fact: Muscle weakness is a symptom of many conditions. A specific diagnosis requires genetic or enzymatic testing.
Treatments and Therapy
Medication-Based Treatments
- Enzyme Replacement Therapy (ERT): This is the cornerstone of treatment. It involves intravenous (IV) infusions of a recombinant form of the GAA enzyme (e.g., alglucosidase alfa, avalglucosidase alfa) to replace the missing enzyme and help clear glycogen accumulation.
Surgical Treatments
- Surgery is not a treatment for Pompe disease itself but may be necessary to manage complications, such as correcting scoliosis or placing a feeding tube (gastrostomy) for those with severe swallowing difficulties.
Physical Therapy and Rehabilitation
- A tailored physical therapy program is crucial to maintain joint flexibility, muscle strength, and range of motion. Respiratory physical therapy can help maintain lung capacity.
Lifestyle and Behavioral Interventions
- Nutritional Support: Working with a dietitian to ensure adequate calorie intake, especially if swallowing is difficult.
- Exercise: Low-impact, non-exhausting exercises like swimming or stationary cycling can be beneficial under professional guidance.
- Energy Conservation: Pacing activities to manage fatigue.
Alternative and Complementary Medicine
- These are not cures but may support well-being. Examples include acupuncture for pain management or yoga for gentle stretching and relaxation. It is essential to discuss all complementary therapies with your healthcare team.
Psychotherapy and Counseling
- Living with a chronic, progressive disease is challenging. Psychological counseling can help patients and families cope with anxiety, depression, and the emotional burden of the diagnosis.
Immunizations and Vaccines
- Staying up-to-date on vaccinations, especially for influenza and pneumonia, is critical to prevent respiratory infections, which can be severe for Pompe patients.
Stem Cell Therapy
- This is considered an experimental approach for Pompe disease and is not a standard treatment. Research is ongoing to determine its potential efficacy and safety.
Gene Therapy
- Gene therapy is a promising area of research that aims to deliver a functional copy of the GAA gene to the patient’s cells, enabling them to produce the missing enzyme on their own. It is currently in clinical trials and not yet widely available.
Top 20 FAQ with Answers on Pompe Disease
- What is Pompe disease?
- It is a rare genetic disorder where glycogen accumulates in muscle cells due to a missing enzyme, causing progressive muscle weakness.
- How is Pompe disease inherited?
- It is inherited in an autosomal recessive pattern, meaning both parents must be carriers of the faulty gene.
- What are the first signs in babies?
- Poor head control, “floppy” muscle tone, feeding difficulties, enlarged tongue, and breathing problems.
- What are the first signs in adults?
- Progressive weakness in legs and hips, difficulty climbing stairs, shortness of breath, and lower back pain.
- Is there a cure for Pompe disease?
- There is no cure, but treatments like Enzyme Replacement Therapy (ERT) can manage the symptoms and slow progression.
- What is the life expectancy for someone with Pompe disease?
- It varies greatly. Without treatment, infantile-onset is often fatal within the first year. With ERT, children are living longer. Life expectancy for late-onset forms has increased significantly with treatment.
- How is Pompe disease diagnosed?
- Through a blood test to measure GAA enzyme activity, confirmed by genetic testing to identify GAA gene mutations.
- Is Pompe disease painful?
- Muscle weakness can lead to discomfort and pain, particularly in the back and joints.
- Can Pompe disease affect the brain?
- Cognitive function is typically not affected. The primary impact is on muscle and nerve cells.
- What is Enzyme Replacement Therapy (ERT)?
- It is an intravenous infusion that provides the body with the missing enzyme to help break down glycogen.
- How often is ERT given?
- It is typically given every other week, with each infusion lasting several hours.
- Can people with Pompe disease have children?
- Yes, but they should seek genetic counseling to understand the risks of passing the disease to their offspring.
- What is the difference between infantile and late-onset Pompe?
- Infantile-onset is more severe, involves the heart, and appears before age 1. Late-onset progresses more slowly, does not typically affect the heart, and can appear from childhood to adulthood.
- Does exercise help with Pompe disease?
- Yes, a supervised, moderate exercise program can help maintain strength and function, but overexertion should be avoided.
- Why is respiratory care so important?
- The muscles that control breathing are often weakened, leading to respiratory failure, which is a major cause of complications.
- Is newborn screening for Pompe disease available?
- Yes, it is part of newborn screening in many countries and an increasing number of U.S. states, allowing for early diagnosis and treatment.
- What specialists are involved in care?
- A multidisciplinary team including neurologists, metabolic geneticists, cardiologists, pulmonologists, physical therapists, and dietitians.
- Can carriers of the gene have symptoms?
- Carriers with one faulty gene copy usually have no symptoms, as one functional gene produces enough enzyme.
- Are there any dietary restrictions?
- No specific diet treats Pompe disease, but a high-protein, low-carbohydrate diet is sometimes recommended to support muscle health. A dietitian should be consulted.
- Where can I find support and more information?
- Reputable organizations include the Pompe Disease News, the Muscular Dystrophy Association (MDA), and the National Organization for Rare Disorders (NORD).
Conclusion
Pompe disease is a severe, progressive neuromuscular disorder caused by the damaging accumulation of glycogen in muscle cells. While it presents significant challenges, the landscape has changed dramatically with the advent of Enzyme Replacement Therapy and the implementation of newborn screening. Early diagnosis and a comprehensive, multidisciplinary treatment approach are paramount to managing symptoms, slowing disease progression, and improving the quality and length of life for those affected. Ongoing research into new therapies, including next-generation ERT and gene therapy, continues to provide hope for even more effective treatments in the future.
Find Trusted Cardiac Hospitals
Compare heart hospitals by city and services — all in one place.
Explore Hospitals
This comprehensive guide on Pompe disease does a great job explaining a rare glycogen storage disorder in terms that are clear and meaningful for both patients and caregivers. The breakdown of how the deficiency of the acid alpha-glucosidase enzyme affects muscle function, especially respiratory and cardiac involvement, helps readers understand the serious impacts of the disease. I appreciate the sections on symptom recognition, diagnostic approaches, and current treatment options, which are especially helpful for those newly facing this condition. Overall, it’s an informative, patient-focused resource that raises awareness and supports informed discussions with healthcare providers.