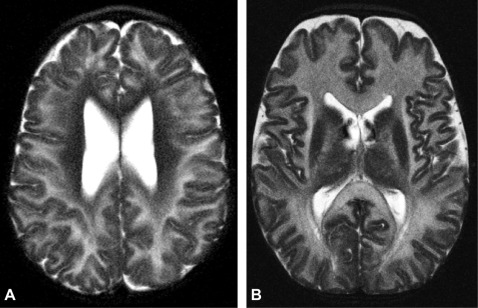
Introduction & Background of Canavan Disease
Canavan Disease is a rare, inherited, and fatal neurodegenerative disorder that belongs to a group of conditions known as leukodystrophies. These disorders are characterized by the imperfect growth or development of the myelin sheath, the fatty covering that acts as an insulator around nerve fibers in the brain.
Myelin is essential for the rapid transmission of nerve impulses. In Canavan Disease, a genetic defect leads to a deficiency of a critical enzyme called aspartoacylase (ASPA). This enzyme is responsible for breaking down a compound called N-acetylaspartic acid (NAA) in the brain. Without functional ASPA, NAA accumulates to toxic levels, which disrupts the process of myelination and leads to the progressive destruction of white matter in the brain. This results in severe neurological impairment and a typically shortened life expectancy.
Causes of Canavan Disease
The primary and sole cause of Canavan Disease is mutations in the ASPA gene, located on chromosome 17. This gene provides the instructions for making the enzyme aspartoacylase.
- Inheritance Pattern: The disease is inherited in an autosomal recessive manner. This means:
- A child must inherit two defective copies of the ASPA gene—one from each parent—to develop the disease.
- Parents of an affected child are carriers, meaning they have one normal copy and one mutated copy of the gene. Carriers do not show symptoms of the disease themselves.
- When two carriers have a child, there is a:
- 25% chance the child will have Canavan Disease.
- 50% chance the child will be an asymptomatic carrier.
- 25% chance the child will be unaffected and not a carrier.
Indications of Canavan Disease
“Indications” often refer to signs that prompt medical investigation. For Canavan Disease, these are typically the early developmental delays observed in infancy. Key indications include:
- Macrocephaly: An abnormally large head size (increased head circumference).
- Significant Hypotonia: Profound lack of muscle tone, often described as “floppiness.”
- Developmental Delay: Failure to meet motor milestones such as head control, rolling over, or sitting up.
- Visual Inattentiveness: The infant does not track objects or make eye contact.
Symptoms of Canavan Disease
Symptoms are progressive and worsen over time. They can be categorized by the stage of the disease.
Infantile Form (Most Common):
- Onset: 3 to 6 months of age.
- Progressive macrocephaly (enlarged head).
- Severe hypotonia (floppiness) that evolves into spasticity (muscle stiffness).
- Loss of previously acquired skills (developmental regression).
- Inability to sit, crawl, or walk.
- Difficulties with feeding and swallowing (dysphagia).
- Reflux and vomiting.
- Seizures.
- Paralysis, blindness, or loss of hearing.
- Sleep disturbances.
Juvenile/Congenital Forms (Rare):
- Much milder symptoms or a later onset.
- Mild developmental delays, speech difficulties, and learning disabilities.
- These forms are exceptionally rare.
Prevention Strategies of Canavan Disease
As a genetic disorder, prevention focuses on genetic counseling and carrier screening.
- Carrier Screening: This is highly recommended for individuals of Ashkenazi Jewish descent, as the carrier rate in this population is significantly higher (about 1 in 40 to 1 in 60). Panel testing can identify the most common ASPA mutations.
- Prenatal Diagnosis: For couples who are both known carriers, testing can be performed during pregnancy to determine if the fetus is affected. This is done via:
- Chorionic Villus Sampling (CVS) at 10-13 weeks.
- Amniocentesis at 15-20 weeks.
- Preimplantation Genetic Diagnosis (PGD): For couples using in vitro fertilization (IVF), embryos can be screened for the ASPA mutation, and only unaffected embryos are implanted.
Myths and Facts About Canavan Disease
| Myth | Fact |
|---|---|
| Canavan Disease is contagious. | It is a genetic disorder, not an infection. It cannot be spread from person to person. |
| Only people of Jewish descent get it. | While it is most common in Ashkenazi Jews, it can occur in any ethnic group. |
| It is caused by something the mother did during pregnancy. | The disorder is entirely genetic and is present from conception. |
| Children with Canavan Disease can “outgrow” it. | It is a progressive, degenerative disease with no cure. However, management can improve quality of life. |
| Carrier testing is 100% accurate. | While highly accurate, no test is perfect. Screening panels look for the most common mutations, but rare ones may be missed. |
Treatments and Therapy
There is currently no cure for Canavan Disease. Treatment is supportive, palliative, and aims to manage symptoms, maintain comfort, and improve quality of life.
Medication-Based Treatments
- Anticonvulsants: To control seizures (e.g., levetiracetam, valproic acid).
- Medications for Spasticity: Such as baclofen or benzodiazepines.
- Medications for Reflux and Feeding Issues: Such as proton pump inhibitors (e.g., omeprazole).
Surgical Treatments
- Gastrostomy Tube (G-tube): A feeding tube surgically placed in the stomach to ensure adequate nutrition and hydration, especially when swallowing becomes unsafe.
- Intrathecal Baclofen Pump: For severe spasticity, a pump can be implanted to deliver medication directly to the spinal fluid.
Physical Therapy and Rehabilitation
- Physical Therapy: To maintain joint flexibility, prevent contractures, and manage spasticity.
- Occupational Therapy: To assist with adaptive positioning and equipment for daily activities.
- Speech Therapy: To address feeding and swallowing difficulties (even if speech is not developed).
Lifestyle and Behavioral Interventions
- Nutritional Support: Working with a dietitian to ensure optimal caloric and fluid intake, often via a G-tube.
- Positioning and Seating: Using specialized chairs, standers, and mattresses to improve comfort, prevent bedsores, and aid digestion.
- Routine Care: Meticulous attention to respiratory secretions, skin care, and oral hygiene.
Alternative and Complementary Medicine
- Acupuncture/Massage: May be used for comfort and to manage muscle stiffness.
- Music and Sensory Therapy: Can provide stimulation and improve quality of life.
Psychotherapy and Counseling
- Family and Sibling Counseling: Essential for helping family members cope with the emotional, financial, and physical demands of caring for a child with a terminal illness.
- Parental Support Groups: Connecting with other families facing Canavan Disease can provide invaluable emotional support and practical advice.
Immunizations and Vaccines
- Children with Canavan Disease should follow the standard vaccination schedule unless otherwise advised by their neurologist, as they are susceptible to standard childhood illnesses.
Stem Cell Therapy
- This is an experimental area of research. The theory is that stem cells could potentially help remyelinate nerve cells or provide a source of the missing enzyme. It is not yet a proven or standard treatment.
Gene Therapy
- Gene therapy is the most promising avenue for a future cure and is under active investigation. The goal is to deliver a functional copy of the ASPA gene into the brain cells, enabling them to produce the aspartoacylase enzyme. Several clinical trials have been conducted, showing some safety and potential for slowing disease progression, but it is not yet a widely available treatment.
Top 20 FAQ with Answer on Canavan Disease
1. What is Canavan Disease?
It is a rare, fatal genetic disorder that causes progressive damage to nerve cells in the brain due to a deficiency of the enzyme aspartoacylase.
2. What causes it?
Mutations in the ASPA gene, inherited in an autosomal recessive pattern from both parents.
3. What are the first signs?
The earliest signs are usually poor head control, low muscle tone (floppiness), and an unusually large head size (macrocephaly) appearing in the first few months of life.
4. How is it diagnosed?
Through a combination of physical exam, brain imaging (MRI showing abnormal white matter), and a urine test showing high levels of N-acetylaspartic acid (NAA). Genetic testing confirms the diagnosis.
5. Can it be treated?
There is no cure. Treatment focuses on managing symptoms like seizures, spasticity, and feeding difficulties to keep the child comfortable.
6. What is the life expectancy?
For the most common infantile form, life expectancy is typically into childhood or the teenage years, though this can vary.
7. Is it painful for the child?
The disease itself is not typically described as painful, but symptoms like muscle stiffness, reflux, and seizures can cause discomfort, which is actively managed with medication and therapy.
8. Can my other children have it?
If you are both carriers, each pregnancy has a 25% chance of resulting in a child with Canavan Disease.
9. Who is at risk?
Individuals of Ashkenazi Jewish descent are at the highest risk, but it can occur in any ethnicity.
10. How common is it?
It is very rare. In the Ashkenazi Jewish population, the incidence is about 1 in 6,400 to 13,500 births.
11. Is there prenatal testing?
Yes, if both parents are known carriers, CVS or amniocentesis can test the fetus for the disease.
12. Can people be carriers without knowing?
Yes, carriers are completely healthy and asymptomatic.
13. Should I get tested?
Testing is strongly recommended if you have a family history of Canavan Disease or are of Ashkenazi Jewish descent.
14. What is the difference between Canavan and other leukodystrophies?
The underlying genetic cause and the specific biochemical defect (NAA accumulation) are unique to Canavan, though the general effect—white matter destruction—is similar.
15. Can a child with Canavan Disease see or hear?
Vision and hearing often deteriorate over time. Many children become blind and/or hearing impaired.
16. Do they recognize their parents?
While cognitive function is severely impaired, many parents and caregivers believe their children respond to familiar voices and touch in their own way.
17. What is gene therapy for Canavan?
An experimental treatment that aims to deliver a working copy of the ASPA gene to brain cells to allow them to produce the missing enzyme.
18. Where can I find support?
Organizations like the National Tay-Sachs & Allied Diseases Association (NTSAD) and the Canavan Foundation provide resources, support, and research information.
19. What kind of daily care is needed?
Total care is required, including feeding (often via a tube), medication administration, physical therapy, and careful positioning.
20. Is there any hope for a cure?
Research into gene therapy and other innovative treatments provides significant hope for future therapies that could slow, halt, or even reverse the disease process.
Conclusion
Canavan Disease is a devastating diagnosis for any family. Its profound impact on a child’s neurological development underscores the critical importance of carrier screening and genetic counseling, particularly in at-risk populations. While a cure remains elusive, a multidisciplinary approach to care can significantly improve the quality of life for affected children and provide essential support for their families. Ongoing research, especially in the field of gene therapy, continues to be a beacon of hope, striving to transform this fatal disorder into a manageable condition in the future.
Find Trusted Cardiac Hospitals
Compare heart hospitals by city and services — all in one place.
Explore Hospitals
This guide is valuable because it explains Canavan disease in a way families can actually act on—starting with the basics that it’s a rare inherited leukodystrophy (usually autosomal recessive) that affects brain white matter and leads to progressive motor and developmental challenges, often beginning in infancy. It also helps readers understand why early diagnosis matters: genetic testing and supportive assessments can confirm the condition, prevent confusion with other neurological disorders, and allow families to plan care sooner (nutrition support, physical/occupational therapy, seizure management, respiratory care, and regular monitoring). I also appreciate the emphasis on genetic counseling, because knowing carrier status and recurrence risk can guide future family planning and support relatives who may also be carriers. While there is no universal cure today, ongoing research into gene-based and metabolic approaches offers hope, and this kind of clear, compassionate information helps caregivers focus on quality of life, comfort, and practical next steps rather than uncertainty.